

Thermal Validation Services
Mapping of Temperature Controlled Units
To verify the performance of temperature controlled units (TCU or CTU) like incubators, fridges, freezers and ultra-low freezers (-80°C) it is important to start with the preparation of a validation protocol. In many cases the customer provides us the validation. In case the validation protocol is not available we can prepare your protocols based on the FD X 15-140 standard.
According to this standard, the number of loggers is based on the volume of the room and certain limits will be verified for the deviations of cold- and hot spots related to the average temperature in the room. Apart from the registration of the temperature distribution we can perform additional tests such as a power failure test and an open-door tests.

Validation of Sterilizers
Sterilisation is mainly done by using saturated steam at a temperature of 121 °C for at least 15 minutes or 134 °C for at least 3 minutes (EN 285). CMI carries out the necessary temperature and pressure measurements to check that the process specifications are met over the entire load. An empty chamber test must not be omitted in order to check the proper functioning of the autoclave against the requirements stated in EN 285, ISO 17665-1 and ISO 17665-2.

When saturated steam hits the colder surface of the material the steam saturated steam condenses on the surface. The heat that is released during this condensation process is sufficient to kill all living microorganisms on the surface of the material. For this reason, it is very important that the quality of the steam is optimal, i.e:
The steam may hardly contain any impurities
The proportion of non-condensable gases is sufficiently low
The steam must not contain any condensate (wet steam)
The steam may not be overheated.
The team of CMI engineers is able to check the quality of the steam on a regular basis according to the criteria mentioned in the EN 285 standard.
Validation of Low Temperature Sterilizers
A second sterilisation method is the use of hydrogen peroxide, H2O2. This application is suitable for heat-sensitive instruments that are not resistant to high temperatures. Liquid hydrogen peroxide is injected under vacuum into the sterilisation chamber. Through evaporation and diffusion of the gas, all surfaces of the load are reached.

The gas is activated into a plasma with formation of free radicals that kill the micro-organisms present. The temperature and pressure are measured during operation with our Datatrace high precision loggers along with the installation of chemical and biological indicators to validate the proper operation of these Low Temperature Sterilisers against the ISO 14937 standard (pending the new ISO 22441)
CSD – Central Sterilization Department
Different types of instruments are used during surgery of a patient. These instruments must be sterile to prevent the possibility of infections and associated complications. After surgery, the used material undergoes a sterilization process within the hospital (with the exception single use instruments) after which it can be reused for the next surgery on a patient. A number of steps are followed to make an instrument sterile. A dirty instrument that arrives at the Central Sterilization Department (CSD) is pre-cleaned manually and / or mechanically. This is followed by mechanical cleaning and disinfection in a washer disinfector provided for this purpose.
The cleaning and disinfection process of a washer disinfector must meet various requirements set in the ISO 15883 standard and must be qualified annually by an external company. By means of thermometric testing over the entire load we validate whether the temperature meets the predefined specifications for a sterilization cycle on every single location in the load with the Datatrace high precision data loggers. The Datatrace pro software can calculate the A0-value based on the logged data for temperature. This value indicates whether a sufficient degree of disinfection has been achieved.
Additional to thermometric testing, cleaning tests are performed using indicators and swab tests. After all, a sufficient degree of disinfection can only take place if the material is free of impurities and blood residues. The washer disinfector uses chemicals for the cleaning process. The dosage is checked at each validation. Once the washing and disinfection process is completed, the material is prepared for the sterilization process. The material, necessary for sterile storage and preservation, is sealed by using containers, non-woven packaging or laminate.


Laminate bags must be sealed by means of a welding machine or sealer. CMI verifies its proper functioning based on the criteria set in EN 868-5 and ISO 11607-2 Standards.
Mapping of Warehouses
Any temperature sensitive product must be stored in a conditioned room where temperature an/or relative humidity must be kept within predefined limits, data loggers are placed over the entire storage area. Special attention should be paid to locations where the probability of temperature excursions are most obvious, for example near a fan, in the vicinity of doors or windows, etc. The logger results from the thermal mapping will be used to determine the cold – and hot spot locations in the warehouse. These locations can be used for monitoring applications with real time alarms.
In consultation with the customer, the locations and the number of probes to be placed will be determined, as well as the duration of the mapping. To assure the conformity under worst case conditions, it is recommended to perform a winter and summer mapping.

In consultation with the customer, the locations and the number of probes to be placed will be determined, as well as the duration of the mapping. To assure the conformity under worst case conditions, it is recommended to perform a winter and summer mapping.

Thermal validation tests:
Determination of cold & hot spot
Temperature distribution
Power failure test
Open door test
Winter mapping
Summer mapping
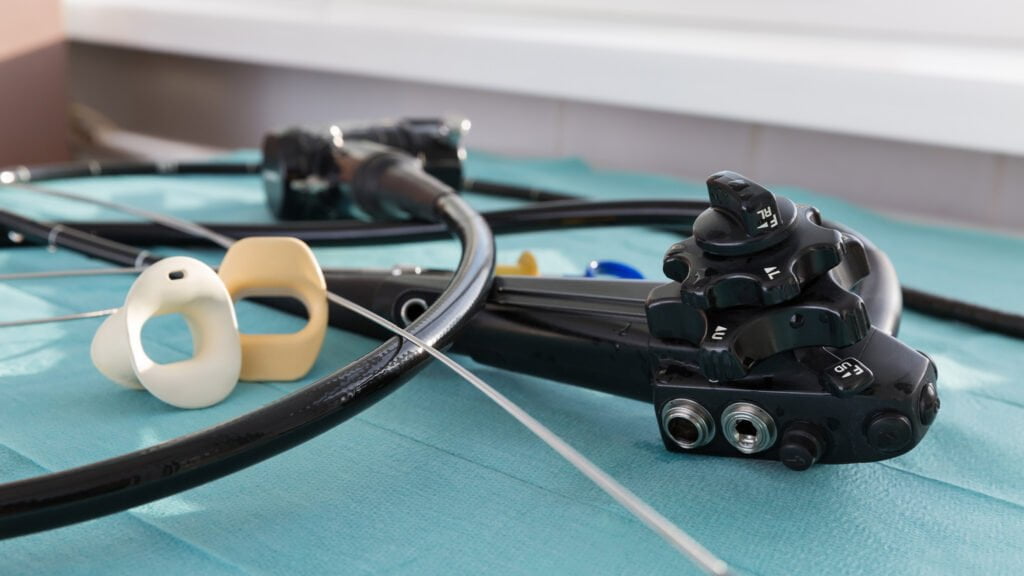
Endoscopies
Endoscopes are used in various applications in internal examinations such as coloscopy, bronchoscopy, gastroscopy, etc. These flexible cameras are reused and need to be thoroughly cleaned and disinfected between examinations. This is achieved by endoscope cleaners. Both the channels and the exterior of the endoscope are rinsed, cleaned and chemically disinfected. Because the material is not resistant to high temperatures, an endoscope cannot be thermally disinfected.
We validate your endoscope cleaners according to the ISO 15883-4 standard and the SFERD handbook. By using a surrogate endoscope, it is possible to measure both temperature and pressure parameters through the channels. Cleaning indicators can be placed on the outside as well on the inside channels to check whether the endoscopes are adequately washed. If channels are not connected, or a channel is blocked, the instrument should generate an alarm. An unconnected channel can lead to insufficient cleaning and disinfection, possibly resulting in contamination. The alarms are checked as well as the product dosage. If required, a microbiological analysis of the last rinse water can be performed. In this way, any biofilm formation in the unit or contamination of the process water can be immediately detected.
Endoscope drying cabinets

Endoscopes are usually not used immediately after the washing and disinfection process, but are stored in a designated storage area. Endoscope drying cabinets have a double function. On the one hand, they blow filtered air through the channels of the endoscope to dry it. On the other hand, the ambient air in a drying cabinet meets at least ISO class 8, which means that an endoscope can be stored for a longer period of time. To put a drying cabinet into use, it must comply with ISO 16442 standard. The CMI verifies this by carrying out, among other things, particle counts, filter tests, microbiological tests.
Execution and reporting
Reports are filled out according to GDocP (Good Documentation Practices) and verified by our own internal QA department before the official report is presented to the customer. The report includes all raw data in compliance with Annex 11 (21 CFR part11) , calibration certificates of the equipment used and conclusions / recommendations based on the results of the measurements.
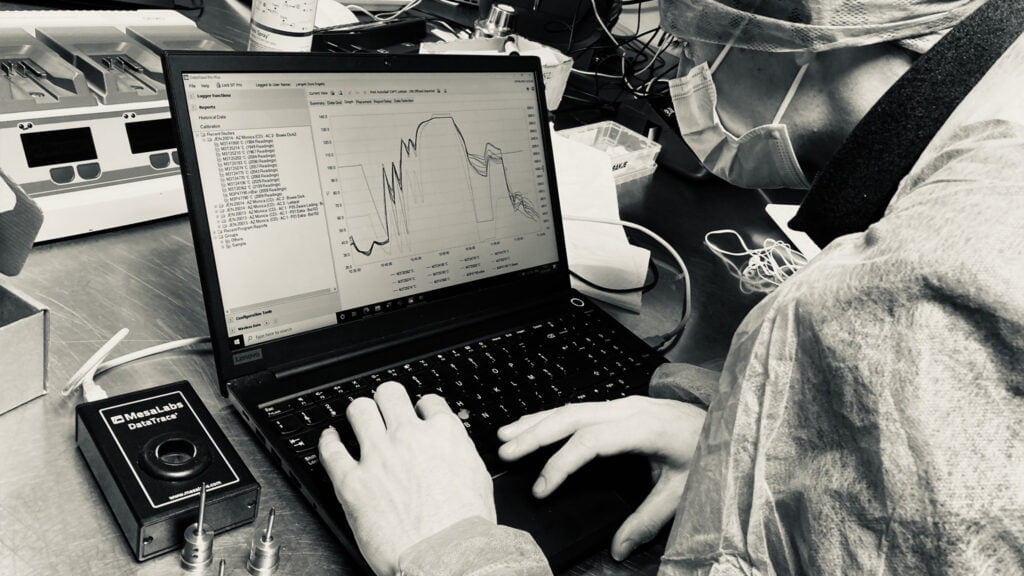
Good Documentation Practice
(GDocP — or GRK for Good Record Keeping) is an essential component of your (and our) overall quality management systems (QMS) and risk management strategies (QRM). All important PASS/FAIL criteria are incorporated in the validation protocol which enables us to qualify the cleanroom or equipment based on these clear criteria.
